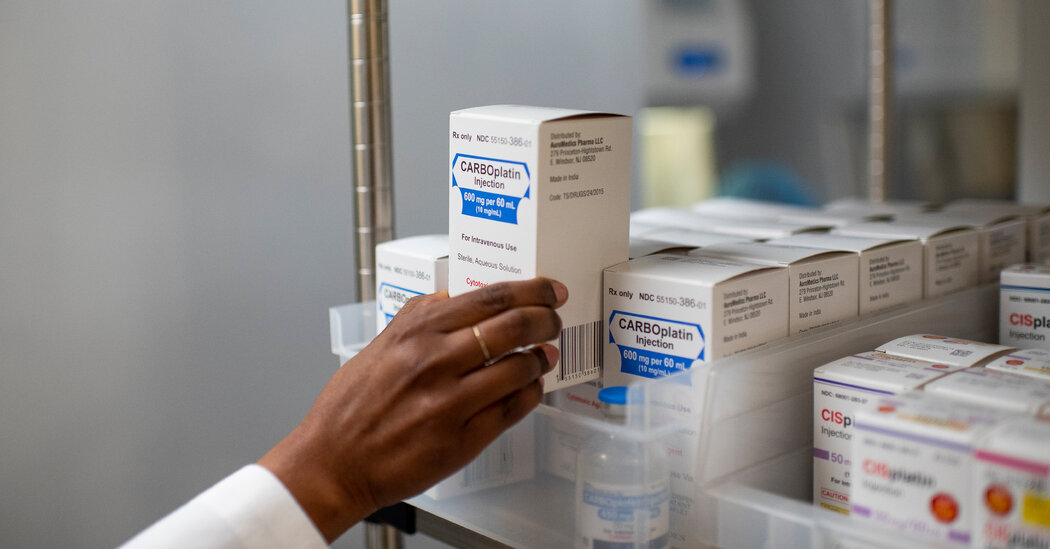
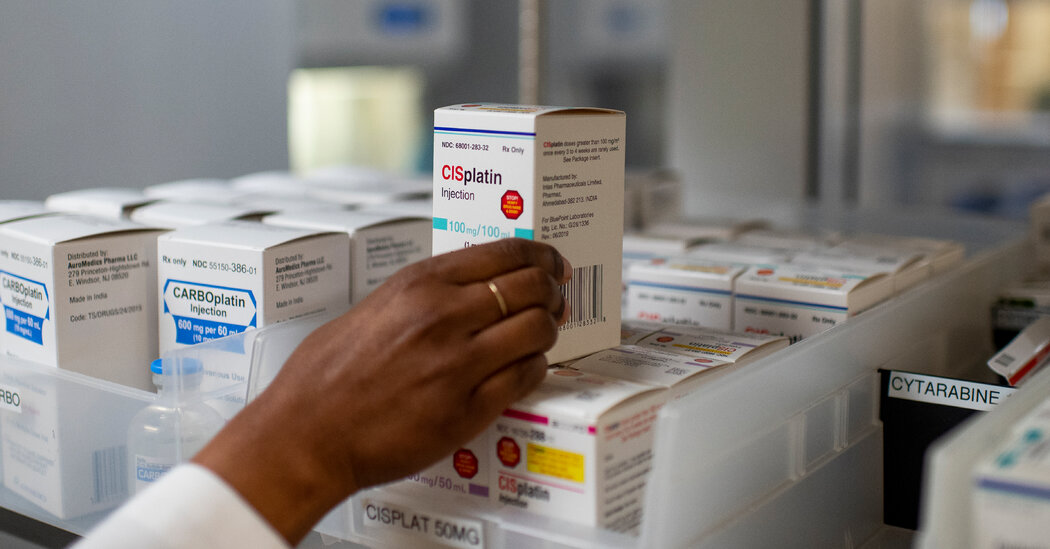
Key drugs have been in scarce supply, revealing a deep crisis in the generic drug industry.Stephanie Scanlan learned about the shortages of basic chemotherapy drugs this spring in the most frightening way. Two of the three drugs typically used to treat her rare bone cancer were too scarce. She would have to go forward without them.Ms. Scanlan, 56, the manager of a busy state office in Tallahassee, Fla., had sought the drugs for months as the cancer spread from her wrist to her rib to her spine. By summer it was clear that her left wrist and hand would need to be amputated.“I’m scared to death,” she said as she faced the surgery. “This is America. Why are we having to choose who we save?”The disruption this year in supplies of key chemotherapy drugs has realized the worst fears of patients — and of the broader health system — because some people with aggressive cancers have been unable to get the treatment they need.Those medications and hundreds of other generic drugs, including amoxicillin to treat infections and fentanyl to quell pain during surgery, remain in short supply. But the deepening crisis has not fostered solutions to improve the delivery of generic drugs, which make up 90 percent of prescriptions in the United States.Dr. Robert Califf, commissioner of the Food and Drug Administration, has outlined changes the agency could make to improve the situation. But he said the root of the problem “is due to economic factors that we don’t control.”“They’re beyond the remit of the F.D.A.,” he said.Senator Ron Wyden, a Democrat of Oregon and chairman of the Senate Finance Committee, agreed. “A substantial portion of these market failures are driven by the consolidation of generic drug purchasing among a small group of very powerful health care middlemen,” he said at a hearing this month.In interviews, more than a dozen current and former executives affiliated with the generic drug industry described many risks that discourage a company from increasing production that might ease the shortages.They said prices were pushed so low that making lifesaving medicines could result in bankruptcy. It’s a system in which more than 200 generic drugmakers compete, at times fiercely, for contracts with three middleman companies that guard the door to a vast number of customers.In some cases, generic drugmakers offer rock-bottom prices to edge out rivals for coveted deals. In other instances, the intermediaries — called group-purchasing organizations — demand lower prices days after signing a contract with a drugmaker.The downward pressure on prices — no doubt often a boon to the pocketbooks of patients and taxpayers — is intense. The group purchasers compete against one another to offer hospitals the lowest-priced products, which intermediary companies say also benefits consumers. They earn fees from drugmakers based on the amount of medications the hospitals buy.“The business model is broken,” said George Zorich, a pharmacist and retired generic drug industry executive. “It’s great for G.P.O.s. Not great for drug manufacturers, not great for patients in some cases.”Ms. Scanlan’s cancer was deemed curable for about 65 percent of patients after cisplatin was added to the cocktail. But during her treatment, Ms. Scanlan got only one dose of a sister drug, carboplatin.Emil Lippe for The New York TimesMany doctors wish they could do more to give cancer patients the medicines they need.“Every clinician I know would be thrilled to pay more money for a reliable supply of a quality drug,” said Dr. Andrew Shuman, a University of Michigan oncology surgeon and expert on drug shortages.In a speech to drug supply intermediaries last month, Dr. Califf exhorted them to “pay more,” saying it would enhance access to medical products and would be “good for business.”Prices fell in recent years for two of the three drugs that Ms. Scanlan was initially offered to treat her cancer. During those years, Intas Pharmaceuticals, a generics giant in India, steadily gained market share as other companies left, according to data from the U.S. Pharmacopeia, a nonprofit that tracks drug shortages.But the company had to halt U.S. production to deal with quality issues that the F.D.A. cited after a surprise inspection of one of its sprawling plants in India. Inspectors had discovered quality-control staff workers shredding and throwing acid on key records. The manufacturing shutdown set off a supply shock in February that would be felt nationwide.Nearly every major U.S. cancer center reported in surveys that they faced chemotherapy shortfalls last spring and summer. One survey released in August found that nearly 60 percent of more than 1,000 pharmacy respondents deemed chemotherapy drug shortages “critically impactful.”Intas recently resumed production, but the F.D.A. still lists the drugs as being in short supply. Major cancer centers report that the shortages are easing, though concerns persist about stock in rural areas.The scarce drugs are cheap and essential and revolutionized their field decades ago, for the first time curing some patients with testicular, lung, ovarian, pancreatic and breast cancers, oncologists say.Ms. Scanlan’s cancer, called osteosarcoma, was deemed curable for about 65 percent of patients after cisplatin was added to the cocktail regimen in the 1970s.Ms. Scanlan’s medical records outline her care. For treatment in the spring and summer, she received only one infusion in March of a sister drug, carboplatin, at University of Florida Shands Hospital in Gainesville.As months passed, Ms. Scanlan’s cancer spread deeper into her bones. She was referred to Tallahassee Memorial Hospital, which, because of the shortages, treated her with one chemotherapy drug. The center then referred Ms. Scanlan to the Mayo Clinic site in Jacksonville in April, according to her medical records.Ms. Scanlan, with her dog, Rosie. “This is America,” Ms. Scanlan said. “Why are we having to choose who we save?”Mark Wallheiser for The New York TimesYet even at the gleaming Florida outpost of the elite medical system, Ms. Scanlan could not get her chemo treatments.By May, she was facing surgery, but might have been eligible to have her wrist repaired rather than amputated. Notes in her records by her Mayo oncology surgeon, Dr. Courtney Sherman, said it would depend on how Ms. Scanlan responded to treatment, though “she is not receiving standard chemotherapy given shortages.”In May and June, both Ms. Scanlan and Dr. Sherman pressed Dr. Steven Attia, a Mayo oncologist, to order the infusions. Ms. Scanlan emailed Dr. Attia: “One question, does Mayo not have the chemo that I actually need?”Dr. Attia declined requests for comment. Samiha Khanna, a Mayo spokeswoman, denied that its site in Jacksonville experienced a cancer drug shortage and confirmed that Mayo did not administer chemotherapy to Ms. Scanlan. Ms. Khanna also referred questions back to Tallahassee Memorial.A market transformedOver the course of his career in the generic drug field, Jeff Herzfeld, a pharmacist and former generics executive who works as a consultant, watched it morph from a field with modest profits to one that is cutthroat.At first it seemed no one was going to make high profits in the generic industry. As drug patents expired, companies entered the market and won customers by offering low prices.But the field of customers began to shrink about 15 years ago. Intermediary companies realized that they could organize hospitals to wield their mass-buying power to get even lower prices.Those intermediaries, or G.P.O.s, charged fees to drugmakers that could access a vast swath of customers. The G.P.O.s competed with one another for hospital clients — enticing them with the lowest prices.The competition stiffened as generic drugmakers vied for each huge deal, emerging victorious if they came in with the lowest price. “They had a winner-take-all approach,” Dr. Herzfeld said.Big deals also came with tough contract terms. One allowed the G.P.O.s to return to the generic drugmaker days after a deal with an ultimatum: Lower the price more or lose the contract. It could happen repeatedly. “You don’t have a lot of room for error,” Dr. Herzfeld said.Generic drug executives said common contract terms deterred them from helping out in a shortage. If they fail to supply promised medications, they can face hefty fines. Yet if they produce more drugs than hospitals buy, they are left with a hole in their balance sheet.Intas Pharmaceuticals, a generics giant in India, steadily gained market share as other companies left the market.Sam Panthaky/Agence France-Presse — Getty ImagesThese routine contract clauses “really penalize or punish” generic drugmakers, said David Gaugh of the Association for Accessible Medicines, which represents the generics industry.Todd Ebert, president of the Healthcare Supply Chain Association, which represents G.P.O.s, disputed those views, arguing that some generic drugmakers offered very low “predatory” prices to force competitors out of the business.Without knowing the cost of producing the drugs, companies cannot be certain if a price is a bargain — or a tactic to hobble the competition, he said. Vizient, a major group purchaser, referred comment to Mr. Ebert.Jessica Daley, a supply chain vice president with Premier, a leading drug-purchasing company, said the company strove to foster healthy markets and wanted “reasonable prices that support supply resiliency and protect patient care.”Aside from the group purchasers’ terms, generic drugmakers also point to other costs they face, including long lists of fees they pay companies that ship drugs from drug factories to hospitals.The current drug shortages have exposed the pressures on the generic market, and the scarcity of cancer treatments has put the spotlight on the troubled growth of Intas Pharmaceuticals in India. It produced two chemo therapies that Ms. Scanlan was to receive early on.Its market share for one of the drugs, methotrexate, which is also used in pediatric cancers and rheumatoid arthritis, grew to 35 percent last year from about 7 percent in 2018, according to the U.S. Pharmacopeia. The data shows the price per dose also fell, to $20 in 2022 from about $25 in 2018.Prices also fell during those years for carboplatin and cisplatin, which tumbled to $15 a dose. Intas’ market share grew, particularly for cisplatin, to 62 percent of the U.S. supply in 2022 from 24 percent in 2018.Ms. Scanlan’s husband, Joe Carr, left, helped wrap her amputated arm in the bathroom.Mark Wallheiser for The New York TimesNot ‘a first-world nation’Dr. Julie Gralow, chief medical officer of the American Society of Clinical Oncology, discovered signs of stockpiling in some health systems as early as February when the F.D.A. first announced the shortage, while shelves were empty at other health centers.“We’re calling it a maldistribution based on who has access — who can afford to create a little stockpile at their site,” Dr. Gralow said.By May her group and others relied on established tenets of bioethics to help cancer centers decide which patients should get scarce treatments, favoring patients with a shot at a cure over those staving off death. Dr. Gralow said researchers were beginning to study whether the chemo shortages are affecting patient survival. Results could take years.The emotional impact has varied widely. Some people with cancer were too focused on paying rent or feeding a family to fight for the medications they desperately needed, said Danielle Saff, a social worker with CancerCare, a nonprofit that supports patients.Others, like Lucia Buttaro, 60, a professor at Fordham University, were furious. She did not get her prescribed carboplatin for a reoccurrence of ovarian cancer in May or June, even though cancer was spreading in her lungs.“In my opinion, we don’t qualify as a first-world nation if you can’t get what you need,” she said.In the case of Ms. Scanlan in Florida, because her cancer was rare, invasive and advanced rapidly, it remains unclear whether the shortages played a role.Still, cancer experts expressed concerns that she had not received standard chemotherapy cocktail regimens before her amputation in September.Failure to use the three “modern miracle” generic chemotherapies for osteosarcoma patients “is a real problem,” said Dr. Lee Cranmer, a sarcoma expert at Fred Hutch Cancer Center in Seattle, who was not involved in Ms. Scanlan’s treatment.She has since received radiation. Last month, she learned the cancer already in her rib and spine had not spread further. Although her new care team at Moffitt Cancer Center in Tampa recently recommended palliative care, she said she felt defeated and terrified.The shortages took a toll, she said, adding: “I can’t help but think about what if something different happened from the beginning.” Ellen Gabler contributed reporting.
Read more →